A quality-by-design approach defines a range of potential viral contaminants of source materials and can be used to effectively achieve viral clearance.
Oct 1, 2014
BioPharm International
Volume 27, Issue 10, pp. 40-47
 The biotech industry has begun to implement a quality-by-design (QbD) approach to ensure the quality of biopharmaceuticals. Using this approach, products are developed with a thorough understanding of how the raw materials and manufacturing processes impact the eventual clinical performance of the final drug product. QbD functions across the lifecycle of the product and integrates sound science with risk management. This approach uses the concepts laid out in the International Conference on Harmonisation of Technical Requirements of Pharmaceuticals for Human Use (ICH) guidelines, Q8 Pharmaceutical Development, Q9 Quality Risk Management, and Q10 Pharmaceutical Quality System.
The biotech industry has begun to implement a quality-by-design (QbD) approach to ensure the quality of biopharmaceuticals. Using this approach, products are developed with a thorough understanding of how the raw materials and manufacturing processes impact the eventual clinical performance of the final drug product. QbD functions across the lifecycle of the product and integrates sound science with risk management. This approach uses the concepts laid out in the International Conference on Harmonisation of Technical Requirements of Pharmaceuticals for Human Use (ICH) guidelines, Q8 Pharmaceutical Development, Q9 Quality Risk Management, and Q10 Pharmaceutical Quality System.
A QbD approach to drug development starts with the definition of critical quality attributes (CQA) that the final drug product will possess, and then outlines the development path to achieve these goals (Figure 1). Raw materials are selected with an understanding of their variability and how this variability will impact the manufacturing process. Risk analysis and scientific characterization identify those components that will have the biggest effect on process consistency and CQA.
Identification of a multivariate design space in which data support the impact that multidimensional process parameter ranges will have on critical quality parameters enables process consistency. The viral safety of a product is an important CQA, and the following is an overview of how QbD can be applied to both the selection and testing of raw materials for adventitious viruses as well as the assurance that the manufacturing process can provide effective and consistent viral clearance.
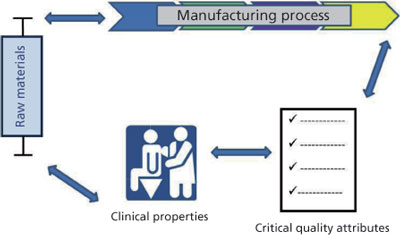 Figure 1: A quality-by-design approach to a product involves an understanding of variability in raw materials and how it impacts the manufacturing process, the relationship of the manufacturing process to the product’s critical quality attributes (CQAs), and how those CQAs impact the product’s function in the clinic.
Figure 1: A quality-by-design approach to a product involves an understanding of variability in raw materials and how it impacts the manufacturing process, the relationship of the manufacturing process to the product’s critical quality attributes (CQAs), and how those CQAs impact the product’s function in the clinic.
QbD in the Viral Safety of Raw Materials
A wide variety of biological raw materials can be used in a biopharmaceutical process, ranging from production cells to buffer components to excipients. These materials can be complex, such as animal serum, or purified products, such as porcine-derived trypsin or a recombinant-affinity chromatography ligand. While the quality of these biological raw materials may significantly impact the consistency of the process or the quality of the final product, the viral safety of raw materials is an important CQA.
Recent and historic contamination events demonstrate the importance of the viral safety of raw materials. Porcine circovirus (1), murine minute virus (2), Cache Valley virus (3,4), and reovirus type 2 (4) are only a few incidences of contamination that have been reported as a result of impure raw materials. Ensuring safety from such contaminations can involve risk assessments, testing raw materials for viral contaminants, and treating raw materials to reduce the risk of viral contaminants.
Each biological raw material possesses a unique risk for transmission of a contaminating virus, and this risk must be assessed. A number of risk assessment tools exist, such as failure mode effects analysis, that can be used to quantitate the likelihood and severity of the risk. The risk assessment may instruct the sourcing of the raw material. Once the relative risk is established, then testing and other risk mitigation measures can be established.
The variety of potential contaminants for a given raw material should be understood in order for effective testing to be done. The list of potential contaminants for a master cell bank derived from Chinese hamster ovary (CHO) cells with a documented history may result in a defined list of tests, but the potential contaminants for a novel cell substrate or a complex biological raw material may be more difficult to determine. For these situations, use of a new molecular technology such as massively parallel sequencing can be informative and provide a good understanding of the types of viruses that may be present, as well as help identify new or novel contaminants. This testing can also be useful if many raw materials routinely turn up.
Massively parallel sequencing or other new technologies such as PCR with mass spectrometry or virus microarrays can be used to identify a panel of potential contaminants. For instance, massively parallel sequencing has been used to evaluate bovine serum, and this testing helped distinguish bovine adeno-associated virus (BAAV-2) as a new contaminant (5). Once a panel of potential viral contaminants is identified in a raw material, assays such as quantitative polymerase chain reaction (PCR) can be developed for use on a routine basis. Thus, a powerful molecular technology is used to identify a range of potential viral contaminants, but standard assays are used on a routine basis.
The testing of raw materials for adventitious viruses is the foundation of any viral safety strategy. However, there are limitations to testing. Very low levels of viral contamination may be below the assay limit and difficult to detect. The volume of raw material tested in an assay may be so small that detection of a viral contaminant is beyond the sensitivity of the assay. Adventitious viruses may not be detectable during sampling if the viral contaminant is not homogeneously distributed in the raw material. Another limitation is that the assay may not be capable of detection of a particular contaminant. Only known viruses can be detected in virus-specific assays, and an unknown virus or variant of a known virus may be missed. A more generalized assay, such as the in-vitro virus assay, may miss an unknown virus or viral variant because the assay requires a cell line that is permissive for the viral contaminant to replicate as well as an endpoint assay by which the virus can be detected.
Because of these testing limitations, it is essential that manufacturing processes that are capable of inactivating or removing potential viral contaminants be factored into the quality analysis. Manufacturers of biopharmaceuticals are beginning to implement viral clearance steps in the upstream process for media and raw materials. These steps can include UV-C irradiation (6), gamma irradiation (7), high-temperature short-time treatment (6, 8) and virus reduction filtration (9, 10). These measures may be optimized to reduce small, non-enveloped viruses, such as parvoviruses, that may be difficult to inactivate or remove in the downstream process. Upstream clearance steps may also reduce the viral load that would have to be eliminated by the downstream process. The clearance afforded by these upstream risk mitigation measures can be quantitated using a viral clearance study.
The viral safety of a raw material used in a manufacturing process for a biopharmaceutical can be evaluated using a QbD approach. The use of powerful, new technologies such as massively parallel sequencing can help to identify the range of viral contaminants that might be present in a raw material. From this information, PCR- or infectivity-based assays can be developed for use in routine testing. ICH Q5A, Note for Guidance on Quality of Biotechnological Products: Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Animal Origin, describes how the level of viral contaminants in process load materials can be compared with the viral reduction capacity of the manufacturing process so that an assessment of the level of viruses in a dose of product can be calculated. This will provide the level of viral clearance required to provide a sufficient margin of safety. Use of upstream risk mitigation measures can be implemented for culture media or media components, thus reducing the risk of viral contamination even further. Understanding the range and level of potential viral contaminants of raw materials has the potential to direct the amount of viral clearance required by the manufacturing process. These factors form the basis of a product that has a high level of viral safety (Figure 2).
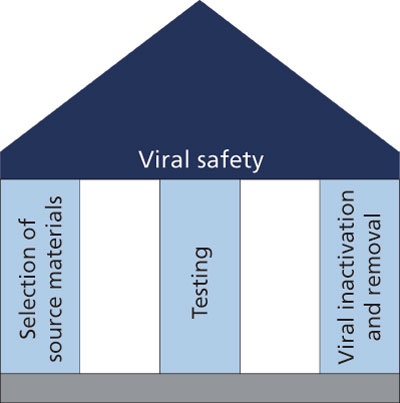 Figure 2: The traditional foundation of viral safety for a biopharmaceutical involves the careful selection of source materials, testing raw materials and process intermediates for viral contaminants, and ensuring that the manufacturing process provides sufficient clearance should a viral contaminant be present. The quality-by-design approach defines a range of potential viral contaminants of source materials so that testing can be designed for relevant contaminants. The viral clearance will apply to a specified design space that allows for changes to process parameters without jeopardizing viral reduction.
Figure 2: The traditional foundation of viral safety for a biopharmaceutical involves the careful selection of source materials, testing raw materials and process intermediates for viral contaminants, and ensuring that the manufacturing process provides sufficient clearance should a viral contaminant be present. The quality-by-design approach defines a range of potential viral contaminants of source materials so that testing can be designed for relevant contaminants. The viral clearance will apply to a specified design space that allows for changes to process parameters without jeopardizing viral reduction.
QbD in Viral Clearance
Regardless of the procedures established to prevent the inadvertent introduction of viral contaminants into a manufacturing process, validating the ability to inactivate and remove viruses will remain an important aspect of achieving the CQA of an acceptable viral safety profile. The amount of viral clearance required will depend on a host of factors, including the nature of the raw materials and the intended patient population. In the broadest sense, this comes down to a minimum number of logs of clearance for different viral classes, based on the risk assessment outlined in ICH Q5A. Because every biopharmaceutical has a unique manufacturing process, the regulatory agencies have thus far avoided providing prescriptive requirements for log reduction, thereby reserving the flexibility necessary in the complex and heterogeneous field of biologic medicines. Therefore, discussions with the regulators early and often in the drug development cycle may help define the details of this CQA and the steps necessary to achieve success.
Multiple unit operations typically contribute to the global viral clearance. The traditional approach to evaluate viral clearance for individual unit operations involves empirical experiments with worst-case conditions when they can be clearly defined, or set-point runs when they cannot. In contrast, a QbD approach involves a more thorough investigation to establish an acceptable “process design space” for each unit operation that achieves a pre-defined goal. Accomplishing this requires a significantly greater upfront investment in development than does the traditional approach. However, the benefits include operational flexibility, as future modifications to the purification method that remain within the process design space are not considered changes and do not require additional viral clearance studies. In addition, the impact of excursions during a production run can be confidently evaluated, reducing clinical and financial risk.
Establishing a process-design space starts with the definition of critical process parameters (CPP) that are likely to have a significant impact on the performance of a given unit operation. This is accomplished through a formal risk analysis that draws on accumulated knowledge about each step, including in-house process development data and publicly available information, such as a database of reported viral clearance compiled by the FDA (11). A parameter should be included as part of a design space if performance (viral clearance) is sensitive to it, excursions are likely (inadvertently or during process improvements), and excursions are not easily detected and corrected in real time. Collectively, the CPPs produce a multidimensional space of possible operating conditions. In a QbD approach, all areas within the space where acceptable performance is achieved are defined through experimentation. A design of experiment approach is typically used because the number of factors and their potential interactions create too many conditions to test practically in a full factorial design.
To date, there are few examples in the literature of this approach, reflecting that this is a relatively new concept, but also reflecting the practical difficulties in executing this strategy. For small companies with limited pipelines and resources, the extensive characterization required to define a viral clearance design space would be difficult to muster, considering the investment required to execute viral clearance studies. Nevertheless, the industry is moving toward a standard requiring a greater degree of understanding and control over manufacturing processes.
Each unit operation is likely to contribute to or impact other CQAs like host cell protein removal for chromatographic steps, for example. Therefore, the definition of an acceptable design space for each unit operation will be dictated by more than one CQA, which also requires characterization. In a low pH hold designed for viral inactivation, for example, a lower pH and longer hold time than some critical values would provide robust inactivation and would be included in the viral clearance design space. Regions within this space may, however, contribute to unacceptable product aggregation. Therefore, the ultimate process design space would be confined by both conditions.
In two recent papers, Genentech (a member of the Roche group) reported on work it has conducted on developing viral clearance process design spaces for two unit operations commonly used in monoclonal antibody production, Protein A and anion exchange chromatography (12, 13). For Protein A chromatography, Genentech found no meaningful effect of the five process parameters tested on the removal of endogenous retroviral particles, thus establishing a large design space for viral safety. It did detect, however, significant differences between feedstocks, limiting the general applicability of its findings to different cell culture harvest substrates. For anion exchange chromatography, Genentech performed experiments with three model viruses, XMuLV, MMV, and SV-40. For all three viruses, anion exchange chromatography achieved robust viral reduction which was largely insensitive to the parameter ranges it tested, again establishing a large design space for this unit operation.
There will be instances when the design-space concept may intersect with the development of modular viral clearance claims. For companies with robust pipelines using closely related manufacturing methods and similar starting materials, some unit operations may be amenable to thorough characterization, developing an operating range that applies generally across molecule type. If all CPPs that vary across a pipeline of molecules could be identified for a low pH hold inactivation step, for example, a cross-platform design space could be defined (14). The success of this approach depends on the ability to identify all CPPs that vary across feedstocks. However, when possible, the resulting cross-platform design space would provide significant efficiencies in the included development programs.
Conclusion
The use of a QbD approach to viral safety requires a thorough understanding of the potential contamination threats in a design space as well as the capacity of the manufacturing process to clear any potential viral contaminants. Understanding how variations in processing parameters impact the capacity of the manufacturing process to remove or inactivate a potential contaminating virus allows the process to provide optimal viral clearance and to predict the impact of process changes on that clearance. For biopharmaceutical products, there will always be a risk of viral contamination. By understanding the nature of potential viral contaminants, implementation of a testing program based on these potential contaminants, and through an understanding of the impact of variations in processing parameters can have on virus clearance, the risk can be managed effectively and viral safety can be achieved.
References
1. J.G. Victoria, C. Wang, M.S. Jones, et al., J. Virol., 84, 6033-6040 (2010).
2. R. L. Garnick, Dev. Biol. Stand. (Basel), 88, 49-56 (1996).
3. R.W. Nims, S.K. Dusing, W-T Hsieh, et al., BioPharm International, 21, 89-96 (2008).
4. A. Kerr, R. Nims, PDA J. Pharm. Sci. Technol. 64, 481-485 (2010).
5. D. Onions and J. Kolman, Biologicals, 38:377-380 (2010).
6. M. Schleh, B. Lawrence, T. Park et al., Am. Pharm Rev., 13, 72-76 (2010).
7. R.W. Nims, G. Gauvin, and M. Plavsic. Biologicals. 39, 370-377 (2011).
8. M. Murphy, G.M. Quesada, and D. Chen, Biologicals. 39 (6) 438-443 (2011).
9. S. Liu, M. Carroll, R. Iverson et al., Biotechnol. Prog. 16, 425-434 (2000).
10. T. Kreil. Proceedings of 17th Planova Workshop (Washington, DC, June 2014).
11. G. Miesegaes, S. Lute, and K. Brorson, Biotechnol. Bioeng. 106, 238-246 (2010).
12. D.M. Strauss T. Cano, N. Cai et al., Biotechnol. Prog. 26, 750-755 (2010).
13. M. Zhang, G.R. Miesegaes, M. Lee et al., Biotechnol. Bioeng. 111, 95-103 (2014).
14. K. Brorson, S. Krejci, K. Lee et al., Biotechnol. Bioeng. 82, 321-329 (2003).
About the Authors
Kathryn Martin Remington, PhD, is a principal scientist, development services John Zehmer, PhD, is a scientist and study director, both with BioReliance by SAFC, Rockville, MD.